A mistura de tensoativos com água, em determinadas proporções, na ausência ou na presença de substâncias lipofílicas pode formar diferentes tipos de agregados, entre os quais agregados polimorfos representados pelas microemulsões (ME) e mesofases liotrópicas - os cristais líquidos (LC), que estão intimamente ligados com a proporção e a natureza dos componentes da mistura.
Foi discutido o papel desses sistemas na incorporação de fármaco com diferentes propriedades físico-químicas, influenciando fortemente a liberação, assim como a biodisponibilidade do fármaco. Aspectos sobre a formação e a caracterização de microemulsões e cristais líquidos também foi discutido.
A análise da literatura indicou que, dependendo da polaridade do fármaco, o efeito da ME ou LC pode ser usado para otimizar o efeito terapêutico por meio do controle da velocidade ou do mecanismo de liberação do fármaco.
Fonte: http://www.scielo.br
31 de outubro de 2011
30 de outubro de 2011
Blog Farmacotécnico é finalista da eleição do Top Blog.
Agora começou a votação do segundo turno.
Votação deste segundo turno vai até 22 de novembro próximo

Graças ao seu voto, nós passamos da primeira fase da eleição para o Top Blog.
Este é um concurso nacional em que os leitores escolhem os blogs que mais gostam.
Com o seu voto ficamos entre os 100 Blogs de Saúde mais votados em todo o país.
Agora começou o segundo e decisivo turno da eleição, aquele que revelará os vencedores finais.
Nessa nova fase, todos os votos dados na primeira fase são anulados. É preciso votar de novo para confirmar sua escolha.
Então, pedimos a você que votou no Blog Farmacotécnico que confirme sua preferência, por favor, votando novamente. E que peça à sua família e aos amigos que votem na gente também.
Para votar, basta clicar no selo acima.
Desde já obrigado por nos trazerem até aqui.
Um abraço forte de toda a equipe do Blog Farmacotécnico.
Votação deste segundo turno vai até 22 de novembro próximo
Graças ao seu voto, nós passamos da primeira fase da eleição para o Top Blog.
Este é um concurso nacional em que os leitores escolhem os blogs que mais gostam.
Com o seu voto ficamos entre os 100 Blogs de Saúde mais votados em todo o país.
Agora começou o segundo e decisivo turno da eleição, aquele que revelará os vencedores finais.
Nessa nova fase, todos os votos dados na primeira fase são anulados. É preciso votar de novo para confirmar sua escolha.
Então, pedimos a você que votou no Blog Farmacotécnico que confirme sua preferência, por favor, votando novamente. E que peça à sua família e aos amigos que votem na gente também.
Para votar, basta clicar no selo acima.
Desde já obrigado por nos trazerem até aqui.
Um abraço forte de toda a equipe do Blog Farmacotécnico.
Avaliação das propriedades de fluxo dos granulados e dissolução de comprimidos de hidroclorotiazida 50 mg obtidos por granulação úmida
O processo de granulação úmida ainda encontra larga aplicabilidade junto à moderna indústria farmacêutica para a produção de comprimidos, pois elimina alguns dos principais problemas atribuídos à compressão direta: a tendência de segregação e as baixas propriedades de fluxo dos pós durante o processo. O presente trabalho avalia e compara através de estudos micromeríticos e análise estatística as características de fluxo de granulados destinados ao desenvolvimento de comprimidos de hidroclorotiazida 50 mg. Foram testados três aglutinantes (pasta de amido e as dispersões aquosas, preparadas a frio, de amido pré-gelatinizado e povidone). Avaliou-se ainda a compatibilidade entre o fármaco e os excipientes empregados, através de estudos termoanalíticos (DSC), e a influência da adição extragranular do superdesintegrante crospovidone nos valores de eficiência de dissolução (%ED) dos comprimidos obtidos. Os granulados obtidos apresentaram propriedades de fluxo e compressibilidade boas a excelentes, caracterizadas por: valores de índice de compressibilidade situados entre 5 e 15; proporções de Hausner inferiores a 1,25 e ângulos de repouso entre 30 e 35º. A adição de crospovidone não incrementou os valores de %ED das formulações desenvolvidas, nas condições experimentais empregadas, ainda que tenha reduzido, de forma pouco significativa, os tempos de desintegração das formulações.
• Os granulados obtidos com os três aglutinantes avaliados apresentaram valores de índice de compressibilidade entre 5 e 15, proporções de Hausner inferiores a 1,25 e ângulos de repouso entre 30 e 35º.
• Comparando-se as propriedades de fluxo dos granulados, antes e após a adição da mistura final, conclui-se:
- Para a formulação baseada em pasta de amido houve melhora estatisticamente significativa das propriedades de fluxo, caracterizada pela redução dos valores de ângulo repouso.
- As propriedades de fluxo das formulações baseadas em povidone (B1 e B2) não foram beneficiadas pela adição dos componentes da mistura final, pois houve redução estatisticamente significativa nos valores de velocidade de escoamento.
- A formulação baseada em povidone apresentou valores de índices de compressibilidade (IC) estatisticamente semelhantes antes e após a adição dos componentes da mistura final. Para as demais formulações houve aumento nos valores de (IC), ainda que este aumento não tenha afetado negativamente as características de fluxo das mesmas.
• A adição do superdesintegrante crospovidone, apesar de reduzir ligeiramente os tempos de desintegração das formulações, não incrementou os valores de eficiência de dissolução (%ED) das formulações desenvolvidas, não sendo, portanto, justificável a sua adição.
• Todas as formulações desenvolvidas apresentaram características de dissolução superiores, caracterizadas pelos valores de %ED estatisticamente superiores, ao do produto referência, nas condições experimentais empregadas.
29 de outubro de 2011
Efeito da Força de Compressão e da Umidade no Perfil de Dissolução de Fármacos

A variabilidade de respostas a um tratamento medicamentoso depende de diversos fatores biofarmacêuticos. Uma forma importante de variação na biodisponibilidade está relacionada com as características físicas e químicas do fármaco, com o processo de fabricação do medicamento e com as características físicas da forma farmacêutica. Neste trabalho, avaliamos a influência da força de compressão e da umidade no perfil de dissolução de uma formulação de comprimidos contendo fenilbutazona.
Resistência mecânica, tempo de desintegração do comprimido e perfil de dissolução do fármaco foram utilizados para avaliar a influência da umidade e da força de compressão. O tempo de desintegração não é alterado com a força de compressão, mas é influenciado pela umidade. O perfil de dissolução foi alterado tanto pela força de compressão como pela umidade. Ambos os fatores podem alterar a biodisponibilidade de fármacos veiculados na forma de comprimidos.
Concluiu-se que a umidade e força de compressão são fatores extrínsecos aos projetos de preparação de medicamentos. Contudo, de acordo com os resultados obtidos neste estudo, podemos concluir que ambos influenciam as propriedades físicas dos comprimidos e o perfil de dissolução do fármaco. Este estudo ressalta a importância de, nos estudos de pré-formulação, definir as condições ideais não só para a fabricação do medicamento, mas também de armazenamento e transporte,uma vez que, como ficou demonstrado, tanto a umidade como a força de compressão podem interferir na taxa de liberação e dissolução de fármacos contidos nas formas farmacêuticas de comprimidos, comprometendo os resultados terapêuticos esperados.
fonte: http://www.unimep.br/phpg/editora/revistaspdf/saude15art06.pdf
28 de outubro de 2011
Análise das cápsulas manipuladas segundo a RDC 67/2007 da ANVISA/ MS para a garantia da qualidade
Este estudo tratou da legislação que norteia o funcionamento das farmácias com
manipulação. A Resolução RDC 67/ANVISA/MS. A fiscalização desses estabelecimentos é de
responsabilidade do Ministério da Saúde, através da ANVISA e seus órgãos regionais, a qual
fez publicar o regulamento técnico que instituía as Boas Práticas de Manipulação em Farmácia
(BPMF). De acordo com esta legislação, a farmácia passa a ser responsável pela qualidade
das preparações que manipula, conserva, dispensa e transporta, e se considera indispensável
o acompanhamento e o controle de todo o processo de obtenção das preparações orais sólidas
(cápsulas), de modo a garantir um produto final de qualidade. O estudo avaliou se a implantação
e a aplicação da resolução garantirão a qualidade no que tange o controle de qualidade
e o monitoramento do medicamento encapsulado, onde se conclui que a norma estudada, não
garante a segurança, eficácia e a qualidade do produto final (medicamento).
Palavras-chave: Farmácia com Manipulação. Resolução. Controle de Qualidade.
Cápsula.
manipulação. A Resolução RDC 67/ANVISA/MS. A fiscalização desses estabelecimentos é de
responsabilidade do Ministério da Saúde, através da ANVISA e seus órgãos regionais, a qual
fez publicar o regulamento técnico que instituía as Boas Práticas de Manipulação em Farmácia
(BPMF). De acordo com esta legislação, a farmácia passa a ser responsável pela qualidade
das preparações que manipula, conserva, dispensa e transporta, e se considera indispensável
o acompanhamento e o controle de todo o processo de obtenção das preparações orais sólidas
(cápsulas), de modo a garantir um produto final de qualidade. O estudo avaliou se a implantação
e a aplicação da resolução garantirão a qualidade no que tange o controle de qualidade
e o monitoramento do medicamento encapsulado, onde se conclui que a norma estudada, não
garante a segurança, eficácia e a qualidade do produto final (medicamento).
Palavras-chave: Farmácia com Manipulação. Resolução. Controle de Qualidade.
Cápsula.
25 de outubro de 2011
Avaliação da estabilidade e atividade antioxidante de uma emulsão base não-iônica contendo resveratrol
Vários são os fatores que podem ocasionar a instabilidade de uma emulsão, destacando-se a oxidação, reação prevenida pelo emprego de antioxidantes. O butil-hidróxi-tolueno (BHT) tem sido um dos antioxidantes sintéticos mais utilizados em formulações cosméticas, porém, a busca da indústria farmacêutica e cosmética pelo emprego de produtos de origem natural tem sido cada vez maior. Visto isso, o objetivo do presente trabalho foi a incorporação do resveratrol, um composto fenólico encontrado principalmente em uvas bem como em vinhos tintos, em uma emulsão base não-iônica para avaliação do perfil de estabilidade e atividade antioxidante em comparação a uma emulsão base não-iônica contendo o BHT. O perfil de estabilidade foi analisado pela observação das características organolépticas, determinação do pH e espalhabilidade, e atividade antioxidante através do teste com o radical livre 2,2-difenil-1-picrilhidrazila (DPPH). Em relação à estabilidade, a altas temperaturas, a emulsão contendo BHT mostrou-se superior à emulsão contendo resveratrol. Pela análise da atividade antioxidante, o resveratrol tanto na sua forma de extrato seco, como quando incorporado na emulsão, demonstrou significativa superioridade em relação ao BHT, podendo ser sua utilização uma alternativa viável em preparações cosméticas, devido ao seu grande potencial antioxidante.
Em relação à análise preliminar da estabilidade da formulação contendo BHT e daquela contendo resveratrol, através da centrifugação das formulações, ambas apresentaram-se estáveis, não sendo necessária a reformulação para a continuidade do estudo.
Os resultados obtidos neste trabalho demonstraram que a EBHT apresenta estabilidade superior em relação à ER, quando ambas são submetidas a altas temperaturas (45° C).
Na avaliação da espalhabilidade de ambas as formulações, nas diversas condições de armazenamento, no decorrer dos 60 dias, a EBHT apresentou maiores valores de espalhabilidade. Cabe ressaltar que a espalhabilidade foi originalmente superior, o que já era esperado, já que a ER apresentava maior teor de sólidos na sua formulação, devido à incorporação do extrato seco contendo resveratrol.
Na avaliação da atividade antioxidante, o emprego do resveratrol justifica-se por ter sido observada sua diferenciada ação na formulação ER em relação à EBHT, que, mesmo após perda de parte de seu potencial antioxidante, quando submetida ao calor, mostrou-se superior ao BHT.
Sendo assim, o desenvolvimento de formulações cosméticas contendo resveratrol como antioxidante mostra-se uma alternativa viável, devido a sua notável superioridade em relação ao antioxidante sintético BHT. Entretanto, é mais aconselhável a utilização de extratos com maiores teores de resveratrol ou da molécula de forma isolada, para que sejam minimizados os efeitos de outras substâncias que constituem os extratos, como por exemplo, excipientes, nas características físico-químicas, bem como na estabilidade da formulação cosmética.
Unitermos: Emulsões/ estabilidade. Antioxidantes. Butil-hidróxi-tolueno. Resveratrol.
http://www.scielo.br/scielo.php24 de outubro de 2011
A Importância da Análise do Peso Médio, Desvio Padrão e Coeficiente de Variação na Manipulação de Medicamentos para a Farmácia com Manipulação
Alexandre Fernandes
Atender às normas de qualidade da Farmacopéia Brasileira IV evita que o estabelecimento que manipula insumos farmacêuticos sofra sanções aplicadas pelo órgão de Vigilância Sanitária. As infrações derivam do não cumprimento dos itens qualificados como Imprescindível (I) e Necessário (N) no Roteiro de Inspeção, constante do Anexo IV da Resolução RDC 33/00, sem prejuízo das ações legais que possam corresponder cada caso.
Verificado o não cumprimento desses itens do Roteiro de Inspeção, deve ser estabelecido um prazo para adequação de acordo com a complexidade das ações corretivas que forem necessárias. Caso haja danos comprovados ao consumidor decorrentes de desvios da qualidade na manipulação de preparações magistrais e oficinais, o responsável estará sujeito às disposições previstas na Legislação Sanitária vigente.
Se a Lei número 6.437/77 art 10, inciso XXIX – por transgredir normas legais e regulamentares destinadas à proteção da saúde – for contrariada, o proprietário do estabelecimento pode levar uma advertência, apreensão, inutilização e/ ou interdição do produto; suspensão de venda e/ou fabricação do produto, cancelamento do registro do produto; interdição parcial ou total do estabelecimento, cancelamento da autorização de funcionamento da empresa, cancelamento do alvará de licenciamento do estabelecimento e proibição de propaganda.
Portanto, para garantir a qualidade do processo, a farmácia deve dispor de um laboratório de controle de qualidade capacitado para a realização de controle em processo e análise da preparação do medicamento, a fim de evitar as penalidades acima.
Passo a passo
Toda farmácia deve manter o Procedimento Operacional Padrão (POP) para a realização do Peso médio e Uniformidade de Doses Unitárias em cápsulas de acordo com o procedimento da Farmacopéia Brasileira IV edição. Para isso, é necessário seguir os seguintes procedimentos:
Peso médio
Com a adoção do desvio padrão e, principalmente, do coeficiente de variação, é possível medir o tamanho do processo. Assim, estabelecerá um indicador para a qualidade do processo de encapsulação do medicamento e verificará a eficiência do manipulador.
Procedimento para cápsulas duras
a) Pesar individualmente 20 cápsulas e determinar o peso médio. Pode-se aceitar a variação dos pesos individuais em relação ao peso médio, conforme indicado na tabela:
Forma farmacêutica
Peso médio ou valor nominal declarado
Limites de variação
Cápsulas duras
Até 300mg
± 10%
Cápsulas duras
Acima de 300 mg
± 7,5%
b) Se uma ou mais cápsulas estiverem fora dos limites indicados, pesar individualmente 20 unidades, remover o conteúdo de cada uma e pesar novamente.
c) Determinar o peso médio do conteúdo pela diferença dos valores individuais obtidos entre a cápsula cheia e vazia.
d) Pode-se tolerar, no máximo, duas unidades fora dos limites especificados na tabela em relação ao peso médio. Porém, nenhuma poderá estar acima ou abaixo do dobro das porcentagens indicadas.
e) Se mais que duas (porém não mais que seis cápsulas) estiverem com variação entre uma e duas vezes o índice da tabela em relação ao peso médio, é necessário determinar o peso do conteúdo em mais 40 unidades e calcular o peso médio das 60.
f) Determinar as diferenças em relação ao novo peso médio. Pode-se tolerar no máximo seis unidades em 60 cápsulas, cuja diferença exceda os limites da tabela em relação ao peso médio, porém, nenhuma cuja diferença exceda o dobro dos mesmos.
É importante que a farmácia mantenha os registros dos cálculos e dos valores dos pesos encontrados.
Desvio padrão e coeficiente de variação
Após a realização do peso médio é importante aplicar os conceitos de desvio padrão e coeficiente de variação, pois somente o peso médio pode proporcionar um resultado final errôneo. O objetivo em se medir o desvio é encontrar uma quantidade que indique a variação em torno da média de um conjunto de medidas.
No Guia Prático da Farmácia Magistral, 2ª edição, página 71 há exemplo que ilustra a explicação acima, o qual será analisado:
Conjunto 1 - Cápsulas contendo: 4mg, 5mg, 6mg, 7mg, 8mg.
Conjunto 2 - Cápsulas contendo: 2mg , 4mg, 6mg, 8mg 10mg .
A média de cada conjunto é igual a seis. Contudo a amplitude dos valores do segundo conjunto é o dobro da amplitude do primeiro, nesse caso o segundo conjunto varia duas vezes mais que o primeiro.
a) Desvio padrão:
Calcular o desvio padrão pela fórmula:
Em que:
s = desvio padrão da amostra;
X = média dos valores obtidos nas unidades testadas;
N = número de unidades testadas;
x1; x2; x3 ..............xn = valores individuais xi das unidades testadas;
n –1 = número de cápsulas testadas menos um. Exemplo: 20 cápsulas testadas: n -1 = 19.
De acordo com a literatura técnica farmacêutica e farmácia galênica, recomenda-se adotar como tolerância para o desvio padrão o valor de 5%.
b) Coeficiente de variação:
Outro conceito é o Coeficiente de Variação (CV), também chamado de estimativa do desvio padrão relativo. É utilizado para expressar a relação percentual da estimativa do desvio padrão com a média dos valores obtidos
s = desvio padrão;
X= média dos valores obtidos.
Tecnologia brasileira
Existe no mercado o Processador Estatístico SP1000, voltado para a indústria cosmética, laboratório químico e farmácia de manipulação, que obtém o peso médio e o desvio padrão relativo, garantindo, assim, a qualidade do encapsulamento. Esse equipamento executa todo o processamento estatístico de um determinado conjunto de amostras e gera um relatório com as seguintes informações:
a) De captura de dados
------------------------------
Processador Estatístico SP1000
Lote: GEHAKA01 Data: 15/07/04
------------------------------
Amostra Peso Hora
1 2,938 10h25
2 2,956 10h26
3 2,911 10h27
4 2,881 10h28
5 2,896 10h29
6 2,943 10h30
7 2,945 10h31
8 2,873 10h32
9 2,943 10h33
10 2,944 10h34
------------------------------
b) Dos resultados finais:
-------- Resultados ---------
N = 10
Máximo = 2,956 g
Mínimo = 2,873 g
Amplitude = 0,083 g
Variância = 0,000908
Desvio padrão = 0,030140
Erro padrão = 0,009531
------------------------------
Média = 2,923 g
Desvio padrão relativo = 1,031 %
==============================
c) Função contagem de peças, depois dos cálculos anteriores:
==============================
Total peso= 200,030 g
Total cápsulas= 78 cápsulas
==============================
O principal diferencial está na captura do valor de pesagem diretamente da balança e a execução de cálculos estatísticos complexos. Com essa metodologia temos como verificar se os medicamentos que foram encapsulados possuem peso médio dentro do valor esperado e com a medida do desvio padrão relativo temos a indicação da variação de peso entre as cápsulas, se este valor estiver dentro de 5% atende aos requisitos do Food and Drug Administration (FDA).
Na conclusão do processo é possível contar a quantidade de cápsulas e documentar por meio da função “contagem de peças”.
Com este relatório está sendo atendido um método de documentação que garante a adequação às normas de qualidade como ISO, Good Laboratory Practices (GLP), ou Boas Práticas de Laboratório (BPL).
O Processador Estatístico opera acoplado às Balanças de Precisão, garantindo assim mais agilidade, confiabilidade e precisão no controle da qualidade de uniformidade no encapsulamento. O equipamento possui conexão com impressora para registrar o peso de cada cápsula que está sendo analisada de uma forma simples e, assim, gerando um relatório que atende a necessidade de documentar o processo conforme a exigência da Agência Nacional de Vigilância Sanitária (ANVISA).
Referências Bibliográficas
(1) Farmacopéia Brasileira IV edição, capítulo V 1.1 da Parte I do Decreto nº 96.607/88;
(2) Ferreira, A. O. Guia Prático da Farmácia Magistral. 2ª Edição, 2002;
(3) Prista, N. L. Técnica Farmacêutica e Farmácia Galênica, I Vol, 3ª edição.
Retirado de: http://www.racine.com.br/portal-racine/farmacias-e-drogarias/manipulacao-magistral/a-importancia-da-analise-do-peso-medio-desvio-padrao-e-coeficiente-de-variacao-na-manipulacao-de-medicamentos-para-a-farmacia-com-manipulacao
Atender às normas de qualidade da Farmacopéia Brasileira IV evita que o estabelecimento que manipula insumos farmacêuticos sofra sanções aplicadas pelo órgão de Vigilância Sanitária. As infrações derivam do não cumprimento dos itens qualificados como Imprescindível (I) e Necessário (N) no Roteiro de Inspeção, constante do Anexo IV da Resolução RDC 33/00, sem prejuízo das ações legais que possam corresponder cada caso.
Verificado o não cumprimento desses itens do Roteiro de Inspeção, deve ser estabelecido um prazo para adequação de acordo com a complexidade das ações corretivas que forem necessárias. Caso haja danos comprovados ao consumidor decorrentes de desvios da qualidade na manipulação de preparações magistrais e oficinais, o responsável estará sujeito às disposições previstas na Legislação Sanitária vigente.
Se a Lei número 6.437/77 art 10, inciso XXIX – por transgredir normas legais e regulamentares destinadas à proteção da saúde – for contrariada, o proprietário do estabelecimento pode levar uma advertência, apreensão, inutilização e/ ou interdição do produto; suspensão de venda e/ou fabricação do produto, cancelamento do registro do produto; interdição parcial ou total do estabelecimento, cancelamento da autorização de funcionamento da empresa, cancelamento do alvará de licenciamento do estabelecimento e proibição de propaganda.
Portanto, para garantir a qualidade do processo, a farmácia deve dispor de um laboratório de controle de qualidade capacitado para a realização de controle em processo e análise da preparação do medicamento, a fim de evitar as penalidades acima.
Passo a passo
Toda farmácia deve manter o Procedimento Operacional Padrão (POP) para a realização do Peso médio e Uniformidade de Doses Unitárias em cápsulas de acordo com o procedimento da Farmacopéia Brasileira IV edição. Para isso, é necessário seguir os seguintes procedimentos:
Peso médio
Com a adoção do desvio padrão e, principalmente, do coeficiente de variação, é possível medir o tamanho do processo. Assim, estabelecerá um indicador para a qualidade do processo de encapsulação do medicamento e verificará a eficiência do manipulador.
Procedimento para cápsulas duras
a) Pesar individualmente 20 cápsulas e determinar o peso médio. Pode-se aceitar a variação dos pesos individuais em relação ao peso médio, conforme indicado na tabela:
Forma farmacêutica
Peso médio ou valor nominal declarado
Limites de variação
Cápsulas duras
Até 300mg
± 10%
Cápsulas duras
Acima de 300 mg
± 7,5%
b) Se uma ou mais cápsulas estiverem fora dos limites indicados, pesar individualmente 20 unidades, remover o conteúdo de cada uma e pesar novamente.
c) Determinar o peso médio do conteúdo pela diferença dos valores individuais obtidos entre a cápsula cheia e vazia.
d) Pode-se tolerar, no máximo, duas unidades fora dos limites especificados na tabela em relação ao peso médio. Porém, nenhuma poderá estar acima ou abaixo do dobro das porcentagens indicadas.
e) Se mais que duas (porém não mais que seis cápsulas) estiverem com variação entre uma e duas vezes o índice da tabela em relação ao peso médio, é necessário determinar o peso do conteúdo em mais 40 unidades e calcular o peso médio das 60.
f) Determinar as diferenças em relação ao novo peso médio. Pode-se tolerar no máximo seis unidades em 60 cápsulas, cuja diferença exceda os limites da tabela em relação ao peso médio, porém, nenhuma cuja diferença exceda o dobro dos mesmos.
É importante que a farmácia mantenha os registros dos cálculos e dos valores dos pesos encontrados.
Desvio padrão e coeficiente de variação
Após a realização do peso médio é importante aplicar os conceitos de desvio padrão e coeficiente de variação, pois somente o peso médio pode proporcionar um resultado final errôneo. O objetivo em se medir o desvio é encontrar uma quantidade que indique a variação em torno da média de um conjunto de medidas.
No Guia Prático da Farmácia Magistral, 2ª edição, página 71 há exemplo que ilustra a explicação acima, o qual será analisado:
Conjunto 1 - Cápsulas contendo: 4mg, 5mg, 6mg, 7mg, 8mg.
Conjunto 2 - Cápsulas contendo: 2mg , 4mg, 6mg, 8mg 10mg .
A média de cada conjunto é igual a seis. Contudo a amplitude dos valores do segundo conjunto é o dobro da amplitude do primeiro, nesse caso o segundo conjunto varia duas vezes mais que o primeiro.
a) Desvio padrão:
Calcular o desvio padrão pela fórmula:
Em que:
s = desvio padrão da amostra;
X = média dos valores obtidos nas unidades testadas;
N = número de unidades testadas;
x1; x2; x3 ..............xn = valores individuais xi das unidades testadas;
n –1 = número de cápsulas testadas menos um. Exemplo: 20 cápsulas testadas: n -1 = 19.
De acordo com a literatura técnica farmacêutica e farmácia galênica, recomenda-se adotar como tolerância para o desvio padrão o valor de 5%.
b) Coeficiente de variação:
Outro conceito é o Coeficiente de Variação (CV), também chamado de estimativa do desvio padrão relativo. É utilizado para expressar a relação percentual da estimativa do desvio padrão com a média dos valores obtidos
s = desvio padrão;
X= média dos valores obtidos.
Tecnologia brasileira
Existe no mercado o Processador Estatístico SP1000, voltado para a indústria cosmética, laboratório químico e farmácia de manipulação, que obtém o peso médio e o desvio padrão relativo, garantindo, assim, a qualidade do encapsulamento. Esse equipamento executa todo o processamento estatístico de um determinado conjunto de amostras e gera um relatório com as seguintes informações:
a) De captura de dados
------------------------------
Processador Estatístico SP1000
Lote: GEHAKA01 Data: 15/07/04
------------------------------
Amostra Peso Hora
1 2,938 10h25
2 2,956 10h26
3 2,911 10h27
4 2,881 10h28
5 2,896 10h29
6 2,943 10h30
7 2,945 10h31
8 2,873 10h32
9 2,943 10h33
10 2,944 10h34
------------------------------
b) Dos resultados finais:
-------- Resultados ---------
N = 10
Máximo = 2,956 g
Mínimo = 2,873 g
Amplitude = 0,083 g
Variância = 0,000908
Desvio padrão = 0,030140
Erro padrão = 0,009531
------------------------------
Média = 2,923 g
Desvio padrão relativo = 1,031 %
==============================
c) Função contagem de peças, depois dos cálculos anteriores:
==============================
Total peso= 200,030 g
Total cápsulas= 78 cápsulas
==============================
O principal diferencial está na captura do valor de pesagem diretamente da balança e a execução de cálculos estatísticos complexos. Com essa metodologia temos como verificar se os medicamentos que foram encapsulados possuem peso médio dentro do valor esperado e com a medida do desvio padrão relativo temos a indicação da variação de peso entre as cápsulas, se este valor estiver dentro de 5% atende aos requisitos do Food and Drug Administration (FDA).
Na conclusão do processo é possível contar a quantidade de cápsulas e documentar por meio da função “contagem de peças”.
Com este relatório está sendo atendido um método de documentação que garante a adequação às normas de qualidade como ISO, Good Laboratory Practices (GLP), ou Boas Práticas de Laboratório (BPL).
O Processador Estatístico opera acoplado às Balanças de Precisão, garantindo assim mais agilidade, confiabilidade e precisão no controle da qualidade de uniformidade no encapsulamento. O equipamento possui conexão com impressora para registrar o peso de cada cápsula que está sendo analisada de uma forma simples e, assim, gerando um relatório que atende a necessidade de documentar o processo conforme a exigência da Agência Nacional de Vigilância Sanitária (ANVISA).
Referências Bibliográficas
(1) Farmacopéia Brasileira IV edição, capítulo V 1.1 da Parte I do Decreto nº 96.607/88;
(2) Ferreira, A. O. Guia Prático da Farmácia Magistral. 2ª Edição, 2002;
(3) Prista, N. L. Técnica Farmacêutica e Farmácia Galênica, I Vol, 3ª edição.
Retirado de: http://www.racine.com.br/portal-racine/farmacias-e-drogarias/manipulacao-magistral/a-importancia-da-analise-do-peso-medio-desvio-padrao-e-coeficiente-de-variacao-na-manipulacao-de-medicamentos-para-a-farmacia-com-manipulacao
Avaliação das propriedades de fluxo dos granulados e dissolução de
O processo de granulação úmida ainda encontra larga
aplicabilidade junto à moderna indústria farmacêutica para a
produção de comprimidos, pois elimina alguns dos principais
problemas atribuídos à compressão direta: a tendência de
segregação e as baixas propriedades de fluxo dos pós durante o
processo. O trabalho avaliou e comparou através de estudos
micromeríticos e análise estatística as características de fluxo de
granulados destinados ao desenvolvimento de comprimidos de
hidroclorotiazida 50 mg.
http://www.scielo.br/pdf/rbcf/v43n3/a12v43n3.pdf
aplicabilidade junto à moderna indústria farmacêutica para a
produção de comprimidos, pois elimina alguns dos principais
problemas atribuídos à compressão direta: a tendência de
segregação e as baixas propriedades de fluxo dos pós durante o
processo. O trabalho avaliou e comparou através de estudos
micromeríticos e análise estatística as características de fluxo de
granulados destinados ao desenvolvimento de comprimidos de
hidroclorotiazida 50 mg.
http://www.scielo.br/pdf/rbcf/v43n3/a12v43n3.pdf
SECAGEM POR ATOMIZAÇÃO NA INDUSTRIA ALIMENTÍCIA: FUNDAMENTOS E APLICAÇÕES

Os secadores por
nebulização, mais conhecidos por “spray dryers” têm como princípio básico a
maximização da área de troca de calor e massa durante a secagem. Esta técnica pode ser
aplicada a qualquer material bombeável, ou seja, com comportamento líquido como, por
exemplo, pastas, lamas, suspensões e soluções. Um dos fatores primordiais para uma
eficiente secagem está na operação de atomização. Neste trabalho, são apresentados e
discutidos os principais fatores que afetam o processo de secagem por atomização, como as
características da pasta, projeto do equipamento, condições de operação e qualidade do
produto secado. Basicamente, são apresentados os três principais tipos de atomizadores: os
bicos de pressão e duplo fluido (pneumático) e o disco rotativo. Cada um dos tipos de
atomizadores encontra aplicações de acordo com a especificidade do material sendo
processado, sendo diferenciados por faixas de tamanhos de gotículas geradas,
uniformidade, gasto energético e capacidade. Também são discutidas neste texto as
possíveis variações nos modos de operação de secadores por nebulização, como por
exemplo as operações em co-corrente, contra-corrente e de escoamento misto. A
associação de nebulizadores e leitos fluidizados também tem grande importância na
indústria de alimentos pois permite a realização simultânea de operações como secagem e
granulação. São apresentados alguns equipamentos LABMAQ de escala laboratorial a
industrial, e uma breve comparação com outros métodos de secagem de alimentos.
Finalmente, algumas aplicações na área alimentícia são apresentadas, em especial a
secagem e instantaneização de leite.
PALAVRAS CHAVE: aplicações; bicos atomizadores; disco rotativo; nebulização;
variações.
*Trabalho financiado pela Labmaq do Brasil Ltda.
CONCLUSÕES
Os procedimentos de eliminação da umidade dos diversos alimentos não são simples. Para tanto, se devem controlar rigorosamente os princípios físicoquimicos
sobre a ação da água nos alimentos. Durante o processo de secagem, um dos problemas que pode ocorrer é o escurecimento do produto devido a ação das enzimas. Também processos não enzimáticos podem causar o escurecimento de alimentos e outros produtos biológicos, devido a reações de Maillard. Degradações de ativos nutricionais por aquecimento também são importantes efeitos na secagem de alimentos.
A escolha adequada do equipamento de desidratação é fundamental, para a obtenção de produtos finais adequados, de boas características e estáveis quanto à conservação do mesmo.
O secador por nebulização é um equipamento consagrado na secagem de alimentos devido a:
- baixa degradação/alteração de nutrientes, aroma, sabor, cor e etc;
- alta produtividade e capacidade;
- efetivo controle das variáveis;
- alta eficiência energética;
- produz materiais diretamente na forma de pó, com forma, tamanho e
densidades controlados pelas condições de processo;
- capacidade de microencapsulação de aromas.
http://www.fcf.usp.br/Ensino/Graduacao/Disciplinas/Exclusivo/Inserir/Anexos/LinkAnexos/secagem%20de%20materiais.pdf
21 de outubro de 2011
Exaustão nos moinhos de martelos
O sistema de exaustão de um moinho de martelos, em uma instalação de moagem bem desenhada, ajudará a reduzir os custos de energia, controlar o pó e diminuir a pressão interna do moinho. Embora a produção varia com o tipo de moinho e velocidade tangencial da extremidade dos martelos, do tamanho dos furos das telas e do produto que esteja triturando, comparados com moinhos sem sistema de exaustão, estes terão um rendimento de 15 a 40% superior.
O volume do produto entrando no interior do rotor e a alta velocidade dos martelos, criarão uma pressão estática na câmara de moagem e nos equipamentos de transporte posteriores ao moinho de martelos.
Se não se vedam adequadamente as portas de inspeção, a pressão estática positiva, despejará, através dos interstícios, pó ao meio ambiente, que se origina pelo efeito de redução do tamanho das partículas. Além das perdas de produto que isso representa, este pó é um combustível potencial para uma explosão com enormes danos materiais.
20 de outubro de 2011
Formas Farmacêuticas
Forma farmacêutica é a forma final de como um medicamento se apresenta: comprimidos, cápsulas, injetáveis, etc. Normalmente as drogas não são administradas aos pacientes, no seu estado puro ou natural, mas sim como parte de uma formulação, ao lado de uma ou mais substâncias não medicinais que desempenham varias funções farmacêuticas. Esses adjuvantes farmacêuticos têm por finalidade, solubilizar, suspender, espessar, diluir, emulsionar, estabilizar, preservar, colorir e melhorar o sabor da mistura final. Com a finalidade de deixar o fármacoagradável ao paladar e eficiente. Existem diferentes formas de apresentação dos medicamentos:
Sólidas:
Semi-sólidas
Líquidas
Pós são preparações farmacêuticas que se caracteriza pela mistura de fármacos e/ou substâncias química finamente divididas e na forma seca. Alguns pós são destinados ao uso interno e outros ao uso externo. Os pós podem ser administrados sob a forma simples ou serem ponto de partida para outras formas farmacêuticas como papéis e cápsulas.
Grânulos são formas farmacêuticas composta de um pó ou uma mistura de pós umedecidos e submetidos a secagempara produzir grânulos de tamanho desejado.
Cápsulas são formas farmacêuticas sólidas, as quais uma ou mais substâncias medicinais e/ou inertes são acondicionadas em um invólucro à base de gelatina. As cápsulas gelatinosas podem ser duras ou moles. São administradas por via oral e possuem propriedades de desintegrarem-se e dissolverem-se no tubo digestivo.
Tabletes ou comprimidos são formas farmacêuticas sólidas de forma variável, cilíndrica ou discóide, obtidas por compressão de medicamentos mais o excipiente,
Drágea são formas farmacêuticas obtidas pelo revestimento de comprimidos. Para este fim, se utiliza diversas substâncias, como: queratina, ácido esteárico e gelatina endurecida com formaldeído.
Pastilhas são formas sólidas destinadas a se dissolverem lentamente na boca, constituída por grande quantidade de açúcar e mucilagens associadas a princípios medicamentosos.
Supositórios são preparações farmacêuticas sólidas, à base de substância fundível pelo calor natural do corpo, destinado a ser introduzido no reto, gerando amolecimento ou dissolução do fármaco. O excipiente mais usado é a manteiga de cacau (lipossolúvel) junto com a glicerina gelatinada (hidrossolúvel).
Óvulos são formas farmacêuticas obtidas por compressão ou moldagem para aplicação vaginal, onde devem se dissolver para exercerem uma ação local. O excipienteem geral é a glicerina.
Pomadas são preparações de consistência pastosa, destinada ao uso externo.
Cremes são emulsões líquidas viscosas do tipo óleo e água ou água e óleo.
Emplastros forma farmacêutica que se dissolve à temperatura do corpo, aderindo-se à pele. É usado como esparadrapo.
Soluções são preparados líquidos obtidos por dissolução de substâncias químicas em água.
Loções são soluções que impregnam na pele; veículo é aquoso e usado sem fricção. Sua fluidez permite aplicaçãorápida e uniforme sobre uma ampla superfície.
Emulsão é uma forma farmacêutica líquida de aspecto cremoso feito com a mistura de um líquido em óleo. Como agentes emulsionantes utiliza-se a goma arábica e a gelatina.
Suspensão são formas farmacêuticas que contêm partículas finas de substâncias ativa em dispersão relativamente uniforme. Deve ser agitado antes do uso.
Extratos fluidos são soluções hidroalcoólicas de constituintes solúveisde drogas vegetais.
Injeções são preparações estéreis de soluções, emulsões ou suspensões destinadas à administração parenteral.
Embalagens para drágeas, cápsulas, comprimidos, pílulas e pó, supositórios e óvulos.
http://www.webartigos.com/artigos/formas-farmaceuticas/33607/
Sólidas:
Semi-sólidas
Líquidas
Pós são preparações farmacêuticas que se caracteriza pela mistura de fármacos e/ou substâncias química finamente divididas e na forma seca. Alguns pós são destinados ao uso interno e outros ao uso externo. Os pós podem ser administrados sob a forma simples ou serem ponto de partida para outras formas farmacêuticas como papéis e cápsulas.
Grânulos são formas farmacêuticas composta de um pó ou uma mistura de pós umedecidos e submetidos a secagempara produzir grânulos de tamanho desejado.
Cápsulas são formas farmacêuticas sólidas, as quais uma ou mais substâncias medicinais e/ou inertes são acondicionadas em um invólucro à base de gelatina. As cápsulas gelatinosas podem ser duras ou moles. São administradas por via oral e possuem propriedades de desintegrarem-se e dissolverem-se no tubo digestivo.
Tabletes ou comprimidos são formas farmacêuticas sólidas de forma variável, cilíndrica ou discóide, obtidas por compressão de medicamentos mais o excipiente,
Drágea são formas farmacêuticas obtidas pelo revestimento de comprimidos. Para este fim, se utiliza diversas substâncias, como: queratina, ácido esteárico e gelatina endurecida com formaldeído.
Pastilhas são formas sólidas destinadas a se dissolverem lentamente na boca, constituída por grande quantidade de açúcar e mucilagens associadas a princípios medicamentosos.
Supositórios são preparações farmacêuticas sólidas, à base de substância fundível pelo calor natural do corpo, destinado a ser introduzido no reto, gerando amolecimento ou dissolução do fármaco. O excipiente mais usado é a manteiga de cacau (lipossolúvel) junto com a glicerina gelatinada (hidrossolúvel).
Óvulos são formas farmacêuticas obtidas por compressão ou moldagem para aplicação vaginal, onde devem se dissolver para exercerem uma ação local. O excipienteem geral é a glicerina.
Pomadas são preparações de consistência pastosa, destinada ao uso externo.
Cremes são emulsões líquidas viscosas do tipo óleo e água ou água e óleo.
Emplastros forma farmacêutica que se dissolve à temperatura do corpo, aderindo-se à pele. É usado como esparadrapo.
Soluções são preparados líquidos obtidos por dissolução de substâncias químicas em água.
Loções são soluções que impregnam na pele; veículo é aquoso e usado sem fricção. Sua fluidez permite aplicaçãorápida e uniforme sobre uma ampla superfície.
Emulsão é uma forma farmacêutica líquida de aspecto cremoso feito com a mistura de um líquido em óleo. Como agentes emulsionantes utiliza-se a goma arábica e a gelatina.
Suspensão são formas farmacêuticas que contêm partículas finas de substâncias ativa em dispersão relativamente uniforme. Deve ser agitado antes do uso.
Extratos fluidos são soluções hidroalcoólicas de constituintes solúveisde drogas vegetais.
Injeções são preparações estéreis de soluções, emulsões ou suspensões destinadas à administração parenteral.
Embalagens para drágeas, cápsulas, comprimidos, pílulas e pó, supositórios e óvulos.
http://www.webartigos.com/artigos/formas-farmaceuticas/33607/
17 de outubro de 2011
Granulação
Granulados podem representar um produto intermediário decisivo na elaboração de formas farmacêuticas sólidas. O conhecimento das características ligadas às diversas metodologias de produção constitui, portanto, fator necessário no momento de escolha da técnica a ser empregada.
O artigo descreve os principais métodos de granulação, abordando também os princípios de formação e as propriedades dos produtos formados.
http://www.ufrgs.br/farmacia/cadfar/v16n1/pdf/CdF_v16_n1_p13_20_2000.PDF
O artigo descreve os principais métodos de granulação, abordando também os princípios de formação e as propriedades dos produtos formados.
http://www.ufrgs.br/farmacia/cadfar/v16n1/pdf/CdF_v16_n1_p13_20_2000.PDF
Bombas de insulina melhoram o controle glicêmico, segundo estudo
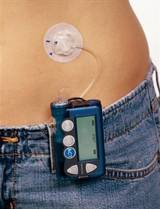 Estudo publicado nos EUA mostra a importância das bombas de insulina (SAP) para o controle glicêmico em paciente com diabetes tipo 1, a terapia tem se mostrado mais eficaz que as que utilizam múltiplas injeções de insulina diariamente.
Estudo publicado nos EUA mostra a importância das bombas de insulina (SAP) para o controle glicêmico em paciente com diabetes tipo 1, a terapia tem se mostrado mais eficaz que as que utilizam múltiplas injeções de insulina diariamente.
O principal benefício é o potencial para reduzir significativamente a os níveis de hemoglobina A1c, por mais tempo que as injeções de insulina mais, sem aumento de hipoglicemia.
Pesquisadores acreditam que o SAP é um passo importante para desenvolver o que é chamado de "sistema de circuito fechado", muitas vezes referido como um "pâncreas artificial". O pâncreas artificial será um dispositivo que pode ler com precisão a glicose no sangue e entregar uma quantidade precisa de insulina automaticamente, da mesma forma que o pâncreas realiza em pessoas sem o diabetes tipo 1.
Alguns pesquisadores sugeriram que, com base em dados de pesquisas realizadas nos EUA, a terapia com bomba de insulina pode não ter um bom custo-efetivo na maioria dos cenários, mas o custo-efetividade poderia melhorar significativamente com os avanços tecnológicos que estão atualmente em desenvolvimento.
"O trabalho continua em direção à integração de plataformas SAP e algoritmos de controle que pode seguramente reduzir a exposição de hipoglicemia e pode fornecer algum dia totalmente um sistema de circuito fechado para aplicação de insulina", acrescentam
Para mais informações acessar o artigo: http://care.diabetesjournals.org/content/early/2011/09/19/dc11-1248.abstract
Fonte:
American Diabetes Association
16 de outubro de 2011
ESTUDO DE DIFERENTES PROCESSOS DE MISTURA DE PÓS-USADOS PARA O PREPARO DE CÁPSULAS EM FARMÁCIAS MAGISTRAIS

Uma operação fundamental para garantir a eficácia dos medicamentos manipulados nas farmácias magistrais é o processo de mistura de pós. Este consiste na distribuição ao acaso das diferentes partículas do sistema, onde cada partícula dos constituintes esteja junto às partículas dos outros. O objetivo do trabalho é avaliar através do teor de princípio ativo os processos de mistura comumente uti lizados nas farmácias de manipulação que são: sacos plásticos, misturadores automáticos e em gral de porcelana. A verificação da qualidade das cápsulas foi feita pela determinação do peso médio, desvio padrão, coeficiente de variação percentual e teste de desintegração. Já a eficiência dos métodos de mistura foi avaliada pela determinação do teor de princípio ativo por espectrofotometria na região do ultravioleta. As fórmulas preparadas pelos diferentes processos de mistura apresentaram boa qualidade. E, em termos de eficiência, os melhores processos de mistura obtidos foram os que utilizaram o misturador automático e o gral de porcelana.
Conclusão do estudo:
Em termos de qualidade, as cápsulas preparadas pelos diferentes processos de mistura estão dentro dos limites aceitáveis. Limites estes especificados pela Farmacopéia Brasileira IV edição em relação ao peso médio, limites de variação, desvio padrão, coeficiente de variação percentual e teste de desintegração. As quatro primeiras variáveis são de extrema importância para garantir a uniformidade de conteúdo e o teste de desintegração afeta diretamente a absorção, biodisponibilidade e portanto a
ação terapêutica. Apesar das cápsulas obtidas através do processo de mistura em saco plástico apresentarem qualidade o mesmo não ocorreu quanto à eficácia terapêutica. Pois, este processo de mistura originou um teor de princípio ativo inferior ao exigido na monografia do fármaco da USP XXIV. Possíveis causas para a baixa porcentagem de liberação do princípio ativo seriam: perdas de pó durante o processo de mistura e aderência do pó as paredes do saco plástico. Através dos resultados obtidos comprova-se que o processo de mistura realizado em saco plástico é ineficaz e inseguro quanto à concentração do fármaco nas cápsulas. O que restringe seu uso em
farmácias magistrais devido à falta de exatidão quanto à homogeneidade da dose. Já os processos de mistura utilizando gral de porcelana e o misturador automático, apresentaram qualidade da forma farmacêutica e eficácia quanto ao teor de princípio ativo. Consideram-se estes dois processos de mistura os mais eficazes e adequados
para serem executados nas farmácias de manipulação, tendo em vista que ambos promoveram uma excelente liberação do fármaco e também por serem menos passíveis de perdas durante o processo de mistura. Os processos de controle de qualidade descritos
devem ser realizados cuidadosamente para que o produto final tenha qualidade e eficácia assegurada. Pois com o crescimento do consumo de medicamentos manipulados, principalmente na produção de cápsulas é de obrigatoriedade do farmacêutico
seguir todos os parâmetros exigidos e preconizados pela legislação vigente.
Quer saber mais clique aqui
15 de outubro de 2011
IMPORTÂNCIA DO CONTROLE DAS CARACTERÍSTICAS
FÍSICO-QUÍMICAS DE INSUMOS FARMACÊUTICOS NA
QUALIFICAÇÃO DE FORNECEDORES
A qualificação de fornecedores é de fundamental importância no âmbito farmacêutico. Além
de cumprir com as boas práticas de fabricação, possuir um rol de empresas que satisfaçam
as necessidades de suprimento de insumos de forma contínua e dentro das especificações
assegura uma melhor eficiência do processo produtivo e, principalmente, a eficácia de um
medicamento. Características de matérias-primas, como distribuição granulométrica, polimorfismo,
morfologia cristalina, dissolução intrínseca, propriedades térmicas e quiralidade
impactam diretamente no processo produtivo e biodisponibilidade de um medicamento. A
adequada definição das características críticas dos insumos que compõem um medicamento
e o uso de técnicas que sejam discriminativas para monitorá-las é uma excelente
ferramenta na avaliação de fornecedores e controle de qualidade.
Palavras-chave: Quiralidade. Polimorfismo. Propriedades Térmicas. Granulometria. Dissolução
Intrínseca.
retirado de: http://www.revistaanalytica.com.br/paginas/artigos-6.pdf
FÍSICO-QUÍMICAS DE INSUMOS FARMACÊUTICOS NA
QUALIFICAÇÃO DE FORNECEDORES
A qualificação de fornecedores é de fundamental importância no âmbito farmacêutico. Além
de cumprir com as boas práticas de fabricação, possuir um rol de empresas que satisfaçam
as necessidades de suprimento de insumos de forma contínua e dentro das especificações
assegura uma melhor eficiência do processo produtivo e, principalmente, a eficácia de um
medicamento. Características de matérias-primas, como distribuição granulométrica, polimorfismo,
morfologia cristalina, dissolução intrínseca, propriedades térmicas e quiralidade
impactam diretamente no processo produtivo e biodisponibilidade de um medicamento. A
adequada definição das características críticas dos insumos que compõem um medicamento
e o uso de técnicas que sejam discriminativas para monitorá-las é uma excelente
ferramenta na avaliação de fornecedores e controle de qualidade.
Palavras-chave: Quiralidade. Polimorfismo. Propriedades Térmicas. Granulometria. Dissolução
Intrínseca.
retirado de: http://www.revistaanalytica.com.br/paginas/artigos-6.pdf
A Granulação de Materiais
A granulação é o processo pelo qual partículas de pó muito finas se aderem entre si para a formação de uma partícula maior, que na realidade são multi-partículas denominadas de grânulos. Dependendo da aplicação desejada, esses grânulos podem se situar em tamanhos que variam de 0,2 mm até 20 mm ou mais. Esse processo é muito utilizado nas indústrias de fertilizantes, farmacêuticas, alimentícias, químicas, siderúrgicas, mineração e produtos cerâmicos. Normalmente, a granulação começa depois de uma mistura, via seca ou via úmida, dos ingredientes pulverizados ou não, de tal forma que esses componentes alcancem uma distribuição uniforme dentro da mistura (homogeneização). A massa granulada, após sofrer um processo de secagem e classificação granulométrica, pode ser utilizada diretamente como produto final (adubos, medicamentos, alimentos, rações para animais etc) ou como uma forma intermediária, dentro do processo industrial, para a obtenção de outros produtos. Por exemplo, uma massa granulada com um certo teor de umidade pode ser compactada dentro de um molde para formar uma pastilha medicamentosa ou então um revestimento cerâmico.
Palavras-chave: granulação, métodos, mecanismos
14 de outubro de 2011
Avaliação das propriedades de fluxo dos granulados e dissolução de comprimidos de hidroclorotiazida 50 mg obtidos por granulação úmida
O processo de granulação úmida ainda encontra larga aplicabilidade junto à moderna indústria farmacêutica para a produção de comprimidos, pois elimina alguns dos principais problemas atribuídos à compressão direta: a tendência de segregação e as baixas propriedades de fluxo dos pós durante o processo. O presente trabalho avalia e compara através de estudos micromeríticos e análise estatística as características de fluxo de granulados destinados ao desenvolvimento de comprimidos de hidroclorotiazida 50 mg. Foram testados três aglutinantes (pasta de amido e as dispersões aquosas, preparadas a frio, de amido pré-gelatinizado e povidone). Avaliou-se ainda a compatibilidade entre o fármaco e os excipientes empregados, através de estudos termoanalíticos (DSC), e a influência da adição extragranular do superdesintegrante crospovidone nos valores de eficiência de dissolução (%ED) dos comprimidos obtidos. Os granulados obtidos apresentaram propriedades de fluxo e compressibilidade boas a excelentes, caracterizadas por: valores de índice de compressibilidade situados entre 5 e 15; proporções de Hausner inferiores a 1,25 e ângulos de repouso entre 30 e 35º. A adição de crospovidone não incrementou os valores de %ED das formulações desenvolvidas, nas condições experimentais empregadas, ainda que tenha reduzido, de forma pouco significativa, os tempos de desintegração das formulações.
Unitermos: Hidroclorotiazida. Granulação úmida. Comprimidos/perfil de dissolução. Micromerítica
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1516-93322007000300012&lng=pt&nrm=iso
13 de outubro de 2011
Uso de Controle Estatístico de Processo (CEP) para validação de processo de Glibenclamida comprimidos
A validação dos processos é um dos requisitos de boas práticas de fabrição. Trata-se de um ato documentado que atesta que qualquer procedimento, processo, equipamento, material, operação, ou sistema realmente conduza aos resultados esperados, assegurando a produção de lotes uniformes que atendam às especificações requeridas pelo produto.
Dos medicamentos administrados oralmente, as formas sólidas são as preferidas. As razões para tal fato é que tanto os comprimidos como as cápsulas, constituem formas farmacêuticas unitárias que permitem a administração do fármaco na dose exata.
Controle Estatístico de Processo (CEP)
O Controle Estatístico do Processo (CEP) pode ser descrito como um conjunto de ferramentas de monitoramento on-line da qualidade. Com tais ferramentas, consegue-se uma descrição detalhada do comportamento do processo, identificando sua variabilidade e possibilitando seu controle ao longo do tempo, através da coleta continuada de dados e da análise e bloqueio de possíveis causas especiais, responsáveis pelas instabilidades do processo em estudo.
METODOLOGIA
Dentre os medicamentos produzidos na unidade industrial em estudo, foi escolhido o produto glibenclamida na forma de comprimidos simples contendo além de 5mg do principio ativo, excipientes com a função de diluentes e lubrificantes. Trata-se de um antiglicemiante consagrado na terapêutica e disponibilizado na rede pública. O critério de escolha do produto, baseou-se no medicamento de mais baixa dose do elenco conforme recomendação da legislação sanitária em vigor para escolha do produto na validação (Brasil,2003). Justifica-se ainda a escolha deste produto, tendo em vista que uma vez que, nestes, as variáveis permitidas pela especificação do produto e pelo processo são mais estreitas e levam a um processo certamente de mais difícil controle.
O processo de produção deste medicamento consiste basicamente de três etapas: Inicialmente é feito ajuste da granulometria dos pós (tamização) dos componentes da formulação seguindo-se a etapa de mistura e compressão.
Foram estudados os dados de 35 lotes produzidos no ano de 2003, cerca de 42.000.000 de unidades produzidas, utilizando dados de peso médio de 10 comprimidos coletados de 1 em 1 hora ao longo do processo a partir de três equipamentos de compressão distintos. O controle do processo é efetuado pelo controle dos pesos dos comprimidos, parâmetro crítico que representa a uniformidade da dose do medicamento, os quais são ajustados para produzir a faixa de 160mg +_7,5%, isto é, os comprimidos devem atender a especificação do produto que permite uma variação de peso dentro da faixa de 148 a 172mg (Brasil, 1998).
Como ferramenta para tratamento dos dados foi utilizado o pacote computacional Statistica Æ (Statsoft) nos módulos: Quality Control Charts e Time Series Analisys/Forecasting.
Foram construídas e avaliados inicialmente, cartas de controle para a média e desvio-padrão geral dos
35 lotes produzidos. Considerando que os equipamentos de compressão possuem capacidades diferentes, as quantidades de dados amostrados no processo foi variável. Na construção das cartas de controle, foi utilizado um valor médio do número de amostras em cada lote. Foram avaliados os desvios padrão dos pesos em função do equipamento de compressão. Dos 35 lotes analisados foram selecionados os lotes que apresentaram maior variabilidade, isto é maiores desvios, para serem analisados isoladamente e verificada a sua performance. A autocorrelação dos dados foi avaliada pela construção dos diagramas da função FAC para os três lotes selecionados. A estabilidade e a conseqünte previsão de causas especiais atuando sobre o processo foi verificada pela aplicação dos testes de não aleatoriedade. Por fim, foi determinado um intervalo de variação para um nível de confiança de 95% das médias dos pesos, bem como, foi determinado o índice Ppk para o processo para todos os lotes estudados.
A alta segurança requerida na validação de processos farmacêuticos pode ser demonstrada pela utilização de técnicas estatísticas. Neste trabalho utilizou-se o controle estatístico de processo para demonstrar a performance de um processo de compressão do produto glibenclamida produzido pelo Laboratório Farmacêutico do Estado de Pernambuco S/A - LAFEPE. Deste trabalho conclui-se que:
- No geral, o processo de compressão dos comprimidos de glibenclamida apresenta estabilidade estatística.
- Causas especiais estiveram presentes em alguns lotes fabricados, reveladas pela aplicação de testes de não aleatoriedade;
- Equipamentos distintos apresentam níveis de variabilidade semelhantes;
- A autocorrelação entre os dados não foi significativa no processo;
- Foi determinado um intervalo de confiança no qual tem-se 95% de chances de se obter pesos que equivalem a uma uniformidade de 100,11 a 100,47%.
- Foi demonstrado, através da determinação do parâmetro Ppk que o processo é capaz de atender as especificações do produto com bastante segurança.
- Trata-se, pois, de uma ferramenta simples na sua aplicação, capaz de permitir fácil compreensão rápidas ações de controle pelo pessoal de operação, rica nos resultados que pode apresentar do comportamento do processo e apta à validação de processos farmacêuticos, de forma retrospectiva e concorrente, e que apresenta a segurança requerida no estabelecimento do “estado de controle” para processos farmacêuticos.
Este artigo foi retirado do site:
http://www.revbrasfarm.org.br/edicoes/pdf/2004/V85_N3_2004/pag_115a119.pdf
10 de outubro de 2011
RDC 52/2011 dispõe sobre a proibição da anfepramona, femproporex, mazindol e controle da sibutramina
Após a anvisa divulgar no dia 4 de outubro a Anvisa decidiu proibir os medicamentos com as substâncias anfepramona, femproporex e mazindol, e aumentar o controle do uso da Sibutramina. Hoje foi publicado no Diário Oficial a resolução RDC 52/2011 que dispõe sobre o assunto.
Os estabelecimentos comerciais tem o prazo de 60 dias para retirarem do mercado os medicamentos proibidos segundo a RDC 52/2011.
A norma da Anvisa também apresenta novas restrições para medicamentos a base de sibutramina. Entre as novidades, está a obrigatoriedade dos profissionais de saúde, empresas detentoras de registro e farmácias e drogarias de notificarem, obrigatoriamente, o Sistema Nacional de Vigilância Sanitária sobre casos de efeitos adversos relacionados ao uso de medicamentos que contém sibutramina.
Além disso, no momento da prescrição, os profissionais de saúde e pacientes deverão assinar um termo de responsabilidade. Esse termo aponta os casos em que o uso desse medicamento é contra-indicado e os riscos aos quais os pacientes que utilizarão medicamentos a base de sibutramina estarão submetidos.
O termo de responsabilidade será preenchido em três vias. Uma via ficará arquivada no prontuário do paciente, uma via será arquivada na farmácia ou drogaria dispensadora e a outra ficará mantida com o paciente.
Além disso, as empresas que possuem registro de medicamentos à base de sibutramina terão 60 dias para encaminharem à Anvisa um plano de farmacovigilância para minimização de riscos de uso do medicamento. Com base nesse plano, a Anvisa reavaliará, em 12 meses, a segurança de uso dos medicamentos a base de sibutramina.
Países discutem falsificação de medicamentos.
Começou nesta quinta-feira (6), no Rio de Janeiro, a 2 ª Reunião Ordinária do Grupo Técnico de Acesso Universal a Medicamentos da União das Nações Sul-Americanas (Unasul), realizada na sede do Pro-Instituto Sul-Americano de Governo em Saúde (Pro-Isags). Promovida em parceria com a Agência Nacional de Vigilância Sanitária (Anvisa) e a Universidade Federal do Rio de Janeiro (UFRJ), o público da reunião são representantes dos 12 países que compõem a Unasul - Argentina, Brasil, Bolívia, Chile, Colômbia, Equador, Guiana, Paraguai, Peru, Suriname, Uruguai e Venezuela. A programação segue até sábado (8), com foco na discussão sobre a necessidade de programar e implantar ações estratégias de combate à falsificação de medicamentos.
Os participantes da 2ª Reunião Ordinária do Grupo Técnico de Acesso Universal a Medicamentos estão no Rio de Janeiro desde segunda-feira (3), pois a estadia no Brasil também incluiu a participação da comitiva na 1ª Oficina sobre os Sistemas de Vigilância Sanitária da América do Sul, também promovida pelo Pro-Isags, Anvisa e UFRJ.
Assim como na reunião técnica, a falsificação de medicamentos também foi debatida durante 1ª Oficina sobre os Sistemas de Vigilância Sanitária. Ontem (5), Bolívia, Uruguai, Colômbia e Brasil expuseram os pontos fortes e fracos de seus sistemas. Nos dois primeiros dias, os participantes conheceram detalhes do Chile, Equador, Argentina, Paraguai, Peru, Suriname e Uruguai.
Diante da predominância do tema sobre falsificação de medicamentos, o coordenador da Unidade de Medicamentos, Tecnologia e Pesquisa da Organização Pan-Americana de Saúde (Opas), Cristhophe Rérat, lembrou que o braço da Organização Mundial da Saúde nas Américas (OMS) vai criar uma ferramenta virtual para respaldar as Vigilâncias Sanitárias Nacionais da região na regulação de preços dos medicamentos e patentes - um espaço para discussão de estudos clínicos e econômicos a fim de ajudar na tomada de decisões na área. A iniciativa é uma parceria com a Agência Nacional de Vigilância Sanitária (Anvisa) e a Secretaria de Ciência, Tecnologia e Insumos Estratégicos, ambas do Ministério da Saúde do Brasil.
O coordenador também lembrou que representantes da Agência Nacional de Vigilância Sanitária (Anvisa), do Ministério da Saúde do Brasil, se reunirão com a diretora-geral da Organização Mundial da Saúde (OMS), Margareth Chan, no Brasil, durante a Conferência Mundial sobre Determinantes Sociais na Saúde, que será realizada de 19 a 21 desse mês, no Rio de Janeiro. Na pauta da conversa, estará o acesso universal a medicamentos e a tratamentos, e do encontro participarão representantes da indústria farmacêutica.
Além da falsificação de remédios, os participantes da oficina discutiram, também, a criação de uma plataforma regional para a vigilância na região, além da avaliação da situação do setor. Portanto, com base nas discussões, os países que compõem a Unasul vão propor ações integradas para fortalecer a Vigilância Sanitária na região.
Documento tratar diretrizes na área de Vigilância Sanitária
Com base nas discussões dos eventos no Rio, um grupo de relatores consolidará um documento que conterá as diretrizes para a área de Vigilância Sanitária na América do Sul. Essas diretrizes visam, em última análise, o acesso a produtos com qualidade, segurança, eficácia e preços razoáveis, sempre de forma a contribuir para a melhoria da saúde dos povos que vivem nessa região do mundo, diz o coordenador-executivo do Pro-Isags, o ex-ministro da Saúde da gestão Lula, José Gomes Temporão. “Esta atividade é uma das primeiras do conjunto de atividades a que o Isags se propõe no âmbito da formação de quadros dirigentes nas melhores práticas na área da gestão da saúde”, afirma.
Para a farmacêutica Consuelo Morros, do Instituto Nacional de Saúde Rafael Rangel, da Venezuela, a experiência de ter participando da oficina é muito enriquecedora, porque os diferentes países da América do Sul estão, pela primeira vez, reunidos para mostrar os respectivos sistemas de Vigilância Sanitária. “De maneira sincera, podemos mostrar quais são as debilidades e fortalezas, para receber dos demais a oferta de intercâmbio”.
Fonte: Ministério da Saúde
Os participantes da 2ª Reunião Ordinária do Grupo Técnico de Acesso Universal a Medicamentos estão no Rio de Janeiro desde segunda-feira (3), pois a estadia no Brasil também incluiu a participação da comitiva na 1ª Oficina sobre os Sistemas de Vigilância Sanitária da América do Sul, também promovida pelo Pro-Isags, Anvisa e UFRJ.
Assim como na reunião técnica, a falsificação de medicamentos também foi debatida durante 1ª Oficina sobre os Sistemas de Vigilância Sanitária. Ontem (5), Bolívia, Uruguai, Colômbia e Brasil expuseram os pontos fortes e fracos de seus sistemas. Nos dois primeiros dias, os participantes conheceram detalhes do Chile, Equador, Argentina, Paraguai, Peru, Suriname e Uruguai.
Diante da predominância do tema sobre falsificação de medicamentos, o coordenador da Unidade de Medicamentos, Tecnologia e Pesquisa da Organização Pan-Americana de Saúde (Opas), Cristhophe Rérat, lembrou que o braço da Organização Mundial da Saúde nas Américas (OMS) vai criar uma ferramenta virtual para respaldar as Vigilâncias Sanitárias Nacionais da região na regulação de preços dos medicamentos e patentes - um espaço para discussão de estudos clínicos e econômicos a fim de ajudar na tomada de decisões na área. A iniciativa é uma parceria com a Agência Nacional de Vigilância Sanitária (Anvisa) e a Secretaria de Ciência, Tecnologia e Insumos Estratégicos, ambas do Ministério da Saúde do Brasil.
O coordenador também lembrou que representantes da Agência Nacional de Vigilância Sanitária (Anvisa), do Ministério da Saúde do Brasil, se reunirão com a diretora-geral da Organização Mundial da Saúde (OMS), Margareth Chan, no Brasil, durante a Conferência Mundial sobre Determinantes Sociais na Saúde, que será realizada de 19 a 21 desse mês, no Rio de Janeiro. Na pauta da conversa, estará o acesso universal a medicamentos e a tratamentos, e do encontro participarão representantes da indústria farmacêutica.
Além da falsificação de remédios, os participantes da oficina discutiram, também, a criação de uma plataforma regional para a vigilância na região, além da avaliação da situação do setor. Portanto, com base nas discussões, os países que compõem a Unasul vão propor ações integradas para fortalecer a Vigilância Sanitária na região.
Documento tratar diretrizes na área de Vigilância Sanitária
Com base nas discussões dos eventos no Rio, um grupo de relatores consolidará um documento que conterá as diretrizes para a área de Vigilância Sanitária na América do Sul. Essas diretrizes visam, em última análise, o acesso a produtos com qualidade, segurança, eficácia e preços razoáveis, sempre de forma a contribuir para a melhoria da saúde dos povos que vivem nessa região do mundo, diz o coordenador-executivo do Pro-Isags, o ex-ministro da Saúde da gestão Lula, José Gomes Temporão. “Esta atividade é uma das primeiras do conjunto de atividades a que o Isags se propõe no âmbito da formação de quadros dirigentes nas melhores práticas na área da gestão da saúde”, afirma.
Para a farmacêutica Consuelo Morros, do Instituto Nacional de Saúde Rafael Rangel, da Venezuela, a experiência de ter participando da oficina é muito enriquecedora, porque os diferentes países da América do Sul estão, pela primeira vez, reunidos para mostrar os respectivos sistemas de Vigilância Sanitária. “De maneira sincera, podemos mostrar quais são as debilidades e fortalezas, para receber dos demais a oferta de intercâmbio”.
Fonte: Ministério da Saúde
9 de outubro de 2011
Vacina da Espanha pode transformar HIV em 'infecção menor'
Pesquisadores da Espanha testaram uma vacina contra o HIV e conseguiram limitar a ameaça do vírus a uma infecção de gravidade menor comparável a herpes em 90% dos voluntários que participaram da pesquisa. A vacina desenvolvida pelo Centro Nacional de Biotecnologia da Espanha se chama MVA-B e foi testada em 30 voluntários saudáveis, 24 receberam o tratamento com a vacina e outros seis receberam um placebo.
Os testes clínicos estão na primeira fase e, além dos 90% dos voluntários terem desenvolvido uma resposta imune ao vírus, 85% deles mantiveram esta resposta durante, pelo menos, um ano. Os resultados da pesquisa foram publicados nas revistas especializadas Vaccine e Journal of Virology.
Treinamento
O sucesso do tratamento tem como base o fato de que o sistema imunológico pode ser treinado para responder a partículas do vírus e células infectadas. Segundo o professor Mariano Esteban, o pesquisador responsável pelo desenvolvimento da vacina, o processo com a MVA-B funciona como "mostrar uma foto do HIV para que (o sistema imunológico) seja capaz de reconhecê-lo se o encontrá-lo no futuro".
As células principais desta experiência são os linfócitos T e B, os "soldados" encarregados de detectar substâncias estranhas no organismo e enviar os sinais necessários para destruí-las. "Nosso organismo está repleto de linfócitos, cada um programado para lutar contra uma patogenia diferente", disse o cientista.
"É preciso submetê-los a um treinamento quando se trata de uma patogenia que não podem vencer naturalmente, como é o caso do HIV", acrescentou. Os linfócitos B produzem os anticorpos que atacam o vírus antes que eles infectem as células. Os exames de sangue dos voluntários feitos na 48ª semana do tratamento revelaram que 72,7% dos voluntários tratados mantiveram os anticorpos específicos contra o HIV.
Os linfócitos T, por sua vez, são encarregados de detectar e destruir as células infectadas. Os exames de sangue realizados na 48ª semana do tratamento, 32 semanas depois da última aplicação da vacina, revelaram que 38,5% desenvolveram um tipo de linfócito T que luta contra o HIV, chamado CD4+. Outros 69,2% tinham desenvolvido outro tipo de linfócito T que luta contra o vírus, chamado CD8+.
Cautela e próximos passos
Os pesquisadores informaram que não ocorreu nenhum efeito adverso e a saúde dos voluntários no teste da vacina não foi afetada. Mas, apesar do otimismo, os médicos envolvidos nos testes recomendam cautela. "Os resultados precisam ser encarados com cautela, já que o tratamento só foi feito em 30 voluntários e, ainda que estimulem uma resposta potente na maioria dos casos, é cedo para prever se as defesas induzidas (pela vacina) vão evitar a infecção", disse o médico Felipe García, responsável pela equipe de pesquisa da vacina no Hospital Clínic, em Barcelona.
O próximo passo da pesquisa espanhola é verificar a eficácia da vacina em pessoas infectadas com o HIV, para verificar se ela realmente reduz a contagem viral. O pesquisador Mariano Esteban demonstra otimismo com as próximas fases. "A vacina MVA-B provou ser tão potente como qualquer outra vacina em estudo atualmente, ou ainda mais", afirmou.
8 de outubro de 2011
Cientistas testam pílula que acaba com a bebedeira
Cientistas estão trabalhando para criar uma pílula capaz de impedir a bebedeira. Com o seu uso será possível beber sem sentir os sintomas da embriaguez como falta de equilíbrio e reflexos lentos. A substância foi testada em ratos que recebiam uma injeção de álcool capaz de derrubar, mas apresentaram reflexos e capacidade de equilíbrio normal. A pesquisa é da Universidade de Adelaide, na Austrália, e foi feita em parceria com pesquisadores dos Estados Unidos.
Segundo reportagem da ABC, a substância utilizada pelos pesquisadores não é nova, ela se chama naloxone e é atualmente aprovada pelo FDA - órgão que regulamenta medicamentos nos
Estados Unidos - para o tratamento de overdose de heroína em humanos. Mas a forma do naloxone utilizada foi levemente alterada para atingir as células da glia, que protegem o cérebro, e não os neurônios.
De acordo com o Daily Mail, os cientistas descobriram que, ao desativar as células do sistema imunológico do cérebro - as células da glia, que ocupam 90% do cérebro e o protegem de infecções como a meningite - é possível ministrar grandes quantidades de álcool sem perceber os efeitos da bebedeira.
Com o bloqueio dos efeitos do álcool no cérebro os pesquisadores acreditam que, além de evitar os micos causados pela ingestão exagerada de álcool, será possível tratar o alcoolismo, limitando a sensação que o consumo desta bebida tem para os alcoólatras. Os cientistas ainda precisam avançar nas pesquisas para começar a fazer testes em humanos, mas acreditam que a pilula pode estar disponível para a venda em três anos.
5 de outubro de 2011
Metade das mulheres sente cólica menstrual em alguma fase da vida
Cerca de 33 milhões de brasileiras sofrem com cólicas menstruais a ponto de impactar sua produtividade no trabalho, segundo estudo do ginecologista César Fernandes publicado na Revista Brasileira de Medicina.
A produtividade delas pode ser reduzida em até 70% nesse período, e 30% das que têm dor se afastam do emprego por pequenos períodos do dia.
Com o objtivo de explicar o que acontece de vez em quando ou até mensalmente no corpo feminino, o Bem Estar convidou o ginecologista José Bento e o proctologista Fábio Atui. Els explicam os sintomas da cólica menstrual e também da intestinal, por que ocorrem e o que é bom comer ou evitar.
Ao longo da vida, uma mulher menstrua cerca de 400 vezes. Quem engravida perde cerca de 20 ciclos de menstruação. entre a gestação e a fase de amamentação. E a cólica apresenta um fator familiar: mães com o problema possivelmente terão filhas com dores.
CÓLICA: Como ocorre: é a contração da musculatura lisa que forma órgãos como estômago, intestinos e útero. A cólica menstrual, comum nas mulheres, pode ser sinal de endometriose, doença que afeta 15% das jovens em idade fértil. Cerca de 50% das mulheres sentem cólicas menstruais em alguma fase da vida. Na menstruação, sempre cai um pouco de sangue na cavidade abdominal, o que acaba soltando o intestino ou causando diarreia. Outras causas da cólica menstrual são: pólipos, miomas, infecções no útero e cistos no ovário.
Jáa cólica intestinal pode ser causada por maus hábitos alimntares ou infecções. Nos dois casos, é preciso prestar atençã na frequencia, intensidades e no quanto as dores atrapalham o dia a dia.
SINAIS DE ALERTA
Cólica menstrual: dores muito intensas antes, durante ou depois do período menstrual, períodos menstruais longos, fluxo abundante, dor durante ou depois das relações sexuais, ao urinar e/ou defecar, infertilidade.
Cólica intestinal: mudança brusca no funcionamento do intestino, alternância constante entre diarreia e intestino preso
DICAS:
Contra cólica menstrual: exercício físico, hábitos alimentares saudáveis e ingestão regular de fibras, bolsa de ága quente, massagem, acupuntura, bebidas quentes(chá ou chocolate).
Alimentos indicados nas crises de cólica intestinal e diarreia: arroz, clado de carne magra, banana-maça, torradas, água e outros líquidos.
Alimentos a evitar: saladas, bagaço de frutas, fibras, café, leite, sucos, frituras e temperos fortes.
As mulheres mais jovens sofrem mais, pois o corpo ainda está se "entendendo" com a produção de prostraglandina, uma espécie de hormônio. E o movimento de contração do útero ocorre para eliminar o endométrio após não haver fecundação.
Como os hormônios contidos nos anticoncepcionais provocam atrofia do endométrio, local de produção da prostaglandina, a pílula é indicada nos casos de cólica para mulheres com vida sexual ativa e que não desejam engravidar.
Tanto para a cólica menstrual como para a intestinal, há o recurso de medicamentos, principalmente antiesoasmódicos, para alívio da dor. Mas nunca se automedique: procure sempre assistência médica.
Segundo José Bento, a cólica pode começar já na primeira ovulação. Entre outros sintomas, pode causar dor de cabeça, náusea, vômito, diarreia e até desmaios. O médico também destacou a importância de anotar os sintomas em um calendário, sobre o qual se deve falar na hora da consulta.
A produtividade delas pode ser reduzida em até 70% nesse período, e 30% das que têm dor se afastam do emprego por pequenos períodos do dia.
Com o objtivo de explicar o que acontece de vez em quando ou até mensalmente no corpo feminino, o Bem Estar convidou o ginecologista José Bento e o proctologista Fábio Atui. Els explicam os sintomas da cólica menstrual e também da intestinal, por que ocorrem e o que é bom comer ou evitar.
Ao longo da vida, uma mulher menstrua cerca de 400 vezes. Quem engravida perde cerca de 20 ciclos de menstruação. entre a gestação e a fase de amamentação. E a cólica apresenta um fator familiar: mães com o problema possivelmente terão filhas com dores.
CÓLICA: Como ocorre: é a contração da musculatura lisa que forma órgãos como estômago, intestinos e útero. A cólica menstrual, comum nas mulheres, pode ser sinal de endometriose, doença que afeta 15% das jovens em idade fértil. Cerca de 50% das mulheres sentem cólicas menstruais em alguma fase da vida. Na menstruação, sempre cai um pouco de sangue na cavidade abdominal, o que acaba soltando o intestino ou causando diarreia. Outras causas da cólica menstrual são: pólipos, miomas, infecções no útero e cistos no ovário.
Jáa cólica intestinal pode ser causada por maus hábitos alimntares ou infecções. Nos dois casos, é preciso prestar atençã na frequencia, intensidades e no quanto as dores atrapalham o dia a dia.
SINAIS DE ALERTA
Cólica menstrual: dores muito intensas antes, durante ou depois do período menstrual, períodos menstruais longos, fluxo abundante, dor durante ou depois das relações sexuais, ao urinar e/ou defecar, infertilidade.
Cólica intestinal: mudança brusca no funcionamento do intestino, alternância constante entre diarreia e intestino preso
DICAS:
Contra cólica menstrual: exercício físico, hábitos alimentares saudáveis e ingestão regular de fibras, bolsa de ága quente, massagem, acupuntura, bebidas quentes(chá ou chocolate).
Alimentos indicados nas crises de cólica intestinal e diarreia: arroz, clado de carne magra, banana-maça, torradas, água e outros líquidos.
Alimentos a evitar: saladas, bagaço de frutas, fibras, café, leite, sucos, frituras e temperos fortes.
As mulheres mais jovens sofrem mais, pois o corpo ainda está se "entendendo" com a produção de prostraglandina, uma espécie de hormônio. E o movimento de contração do útero ocorre para eliminar o endométrio após não haver fecundação.
Como os hormônios contidos nos anticoncepcionais provocam atrofia do endométrio, local de produção da prostaglandina, a pílula é indicada nos casos de cólica para mulheres com vida sexual ativa e que não desejam engravidar.
Tanto para a cólica menstrual como para a intestinal, há o recurso de medicamentos, principalmente antiesoasmódicos, para alívio da dor. Mas nunca se automedique: procure sempre assistência médica.
Segundo José Bento, a cólica pode começar já na primeira ovulação. Entre outros sintomas, pode causar dor de cabeça, náusea, vômito, diarreia e até desmaios. O médico também destacou a importância de anotar os sintomas em um calendário, sobre o qual se deve falar na hora da consulta.
4 de outubro de 2011
Anvisa proibe anfetamínicos e mantém a venda da sibutramina
Medicamentos com Venda Proíbida: Femproporex, mazindol e anfepramona
Medicamento com Venda restrita permitida: Sibutramina
A Diretoria Colegiada da Anvisa decidiu nesta terça-feira (4/10) pela retirada dos medicamentos inibidores de apetite do tipo anfetamínico do mercado e pela manutenção da sibutramina como medicamento para o tratamento da obesidade com a imposição de novas restrições.
De acordo com a decisão dos diretores, os medicamentos femproporex, mazindol e anfepramona terão seus registros cancelados, ficando proibida a sua produção, o comércio, a manipulação e o uso destes produtos. Estes três medicamentos são do grupo denominado inibidores de apetite do tipo anfetamínico.
O relatório apresentado durante a reunião mostra que o uso dos anfetamínicos está baseado na prática clínica , que é o tipo de evidência menos consistente e menos segura segundo os padrões atuais para registro de medicamentos. O texto informa ainda que há evidências de eventos adversos graves, que associadas à ausência de dados confiáveis de segurança justificaram a decisão dos diretores.
Em relação à sibutramina, a decisão da diretoria da Anvisa, por três votos a um, foi de manter o medicamento no mercado com a inclusão de novas exigência e mais restrições para o uso do produto. O relatório apresentado pelo diretor-presidente, Dirceu Barbano, informa que o perfil de segurança da sibutramina é bem identificado e conhecido, o que permite identificar pacientes que podem ter algum ganho a partir do uso da substância. Um das restrições que será estabelecida é a descontinuidade do uso da sibutramina em pacientes que não tiverem resultados após quatro semanas de uso do produto.
No processo de 643 páginas, elaborado ao longo dos últimos oito meses, os técnicos da Anvisa reuniram todos os dados disponíveis de estudos sobre os medicamentos do tipo anfetamínico e a sibutramina.
Leia mais sobre Anvisa proibe anfetamínicos e mantém a venda da sibutramina - Portal Farmacêutico em pfarma.com.br
http://pfarma.com.br/noticia-setor-farmaceutico/legislacao-farmaceutica/764-anvisa-proibe-anfetaminicos-sibrutamina.html
Medicamento com Venda restrita permitida: Sibutramina
A Diretoria Colegiada da Anvisa decidiu nesta terça-feira (4/10) pela retirada dos medicamentos inibidores de apetite do tipo anfetamínico do mercado e pela manutenção da sibutramina como medicamento para o tratamento da obesidade com a imposição de novas restrições.
De acordo com a decisão dos diretores, os medicamentos femproporex, mazindol e anfepramona terão seus registros cancelados, ficando proibida a sua produção, o comércio, a manipulação e o uso destes produtos. Estes três medicamentos são do grupo denominado inibidores de apetite do tipo anfetamínico.
O relatório apresentado durante a reunião mostra que o uso dos anfetamínicos está baseado na prática clínica , que é o tipo de evidência menos consistente e menos segura segundo os padrões atuais para registro de medicamentos. O texto informa ainda que há evidências de eventos adversos graves, que associadas à ausência de dados confiáveis de segurança justificaram a decisão dos diretores.
Em relação à sibutramina, a decisão da diretoria da Anvisa, por três votos a um, foi de manter o medicamento no mercado com a inclusão de novas exigência e mais restrições para o uso do produto. O relatório apresentado pelo diretor-presidente, Dirceu Barbano, informa que o perfil de segurança da sibutramina é bem identificado e conhecido, o que permite identificar pacientes que podem ter algum ganho a partir do uso da substância. Um das restrições que será estabelecida é a descontinuidade do uso da sibutramina em pacientes que não tiverem resultados após quatro semanas de uso do produto.
No processo de 643 páginas, elaborado ao longo dos últimos oito meses, os técnicos da Anvisa reuniram todos os dados disponíveis de estudos sobre os medicamentos do tipo anfetamínico e a sibutramina.
Leia mais sobre Anvisa proibe anfetamínicos e mantém a venda da sibutramina - Portal Farmacêutico em pfarma.com.br
http://pfarma.com.br/noticia-setor-farmaceutico/legislacao-farmaceutica/764-anvisa-proibe-anfetaminicos-sibrutamina.html
2 de outubro de 2011
Polipílula contra hipertensão e colesterol é testada no Brasil
Pacientes com risco cardíaco poderão diminuir o número de remédios diários. Uma pílula que reúne quatro medicamentos para controlar a pressão, baixar o colesterol e prevenir o entupimento de vasos sanguíneos - três fatores que aumentam o risco cardíaco - está sendo testada em diferentes países, incluindo o Brasil, para reduzir a incidência de doenças cardiovasculares.
Aspirina e sinvastatina não oferecem benefício para a hipertensão arterial pulmonar, diz artigo apresentado na conferência internacional da American Thoracic Society
Embora habitualmente usadas para tratar doenças cardíacas, a aspirina e a sinvastatina não mostraram benefícios na hipertensão arterial pulmonar (HAP), uma doença progressiva caracterizada pelo aumento da pressão sanguínea em artérias pulmonares, de acordo com uma nova pesquisa apresentada na conferência internacional da American Thoracic Society, que aconteceu esta semana em Denver, Colorado.
Em estudo financiado pelo U.S. National Institutes of Health, pesquisadores dividiram 65 pacientes em quatro grupos: um recebendo aspirina, outro usando sinvastatina, um grupo tomando ambas as medicações e o último usando placebo.
Surpreendentemente os resultados indicaram que a aspirina e a sinvastatina não mostraram evidências de benefícios clínicos na população estudada, segundo Dr. Steven Kawut, coordenador do estudo e professor de epidemiologia na University of Pennsylvania School of Medicine.
Após receberem as medicações por 6 meses, os pacientes foram avaliados para verificar o quão longe poderiam caminhar em 6 minutos. A distância tendeu a ser menor no grupo que recebeu sinvastatina. Não houve diferenças naqueles que usaram aspirina ou placebo. Estes achados não suportam a rotina de tratamento atual com estas medicações em pacientes com hipertensão arterial pulmonar.
A HAP é uma doença grave, rara e incurável. Ela causa dificuldades respiratórias progressivas, fadiga e vertigens. Pode estar ligada à insuficiência cardíaca e morte.
Os autores concluem que a aspirina e a sinvastatina devem continuar a serem usadas para as indicações clínicas habituais em pacientes com HAP, mas não devem ser administradas para tratar especificamente a hipertensão arterial pulmonar.
É importante lembrar que pesquisas apresentadas em conferências mostram resultados preliminares que precisam ser avaliados em publicações sujeitas à apreciação prévia de avaliadores capacitados.
Fonte: International Conference of American Thoracic Society, 13 a 18 de maio de 2011
Embora habitualmente usadas para tratar doenças cardíacas, a aspirina e a sinvastatina não mostraram benefícios na hipertensão arterial pulmonar (HAP), uma doença progressiva caracterizada pelo aumento da pressão sanguínea em artérias pulmonares, de acordo com uma nova pesquisa apresentada na conferência internacional da American Thoracic Society, que aconteceu esta semana em Denver, Colorado.
Em estudo financiado pelo U.S. National Institutes of Health, pesquisadores dividiram 65 pacientes em quatro grupos: um recebendo aspirina, outro usando sinvastatina, um grupo tomando ambas as medicações e o último usando placebo.
Surpreendentemente os resultados indicaram que a aspirina e a sinvastatina não mostraram evidências de benefícios clínicos na população estudada, segundo Dr. Steven Kawut, coordenador do estudo e professor de epidemiologia na University of Pennsylvania School of Medicine.
Após receberem as medicações por 6 meses, os pacientes foram avaliados para verificar o quão longe poderiam caminhar em 6 minutos. A distância tendeu a ser menor no grupo que recebeu sinvastatina. Não houve diferenças naqueles que usaram aspirina ou placebo. Estes achados não suportam a rotina de tratamento atual com estas medicações em pacientes com hipertensão arterial pulmonar.
A HAP é uma doença grave, rara e incurável. Ela causa dificuldades respiratórias progressivas, fadiga e vertigens. Pode estar ligada à insuficiência cardíaca e morte.
Os autores concluem que a aspirina e a sinvastatina devem continuar a serem usadas para as indicações clínicas habituais em pacientes com HAP, mas não devem ser administradas para tratar especificamente a hipertensão arterial pulmonar.
É importante lembrar que pesquisas apresentadas em conferências mostram resultados preliminares que precisam ser avaliados em publicações sujeitas à apreciação prévia de avaliadores capacitados.
Fonte: International Conference of American Thoracic Society, 13 a 18 de maio de 2011
Assinar:
Postagens (Atom)
Comparação entre técnicas de granulação via úmida: leito fluidizado x alto cisalhamento.
A granulação pode ser realizada por diferentes tecnologias as quais se diferenciam essencialmente nas metodologias utilizadas e,...

-
 Estamos no século XIX. Eu e você somos boticários na cidade, recebemos inúmeras queixas de mães angustiadas com os choros ininterruptos ...
Estamos no século XIX. Eu e você somos boticários na cidade, recebemos inúmeras queixas de mães angustiadas com os choros ininterruptos ... -
Ao contrário do que se pode imaginar, a quantidade de radiação utilizada na Medicina Nuclear é muito pequena. Com isso os tratamentos, proce...
